Protein Catalysis
Return to Ongoing Research TopicsKetosteroid Isomerase Catalysis
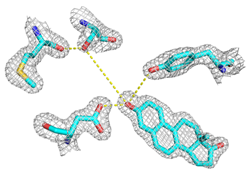 Understanding the mechanisms by which enzymes catalyze chemical reactions with enormous rate enhancements and exquisite specificity is central to biology. And knowledge of the chemical and physical basis of catalysis provides fundamental information that may facilitate the design of novel, artificial enzymes and aid the design of enzyme inhibitors that may act as drugs.
Understanding the mechanisms by which enzymes catalyze chemical reactions with enormous rate enhancements and exquisite specificity is central to biology. And knowledge of the chemical and physical basis of catalysis provides fundamental information that may facilitate the design of novel, artificial enzymes and aid the design of enzyme inhibitors that may act as drugs.
There have been astounding advances in the understanding of enzyme mechanism over the past decades. Nevertheless, fundamental questions remain, and indeed, much of the previous work has helped to bring these critical questions into focus.
Enzymes cannot be described as simply a collection of catalytic amino acids and cofactors. Rather, these groups come together with a precise active site arrangement in a unique, idiosyncratic environment created by the protein scaffold. It is the large number of interactions and this ‘context’ that distinguish enzymes from simple, small molecule catalysts and that must be explored to achieve the next level of understanding of enzymatic catalysis.
The bacterial enzyme ketosteroid isomerase (KSI) provides a remarkably tractable system for in-depth dissection of the mechanisms of catalysis used by enzymes: It catalyzes a simple, well-characterized chemical transformation; it has an established kinetic and thermodynamic reaction framework; it has available transition state analogs to allow structural and spectroscopic probes of active site interactions; it has known, high resolution x-ray crystal structures and is amenable to rapid determination of X-ray structures of new variants and complexes; it catalyzes equilibration of a single substrate/single product reaction, allowing enzyme/substrate complexes to be observed by crystallography and other structural techniques; it is amenable to a panoply of biophysical probes, including NMR and vibrational spectroscopy; and it can be generated semi-synthetically to allow the incorporation of series of unnatural amino acids to systematically and rationally test specific mechanistic proposals.
Some leading papers from the lab in the areas of enzyme mechanism and ketosteroid isomerase are:
- Kraut, D.A., Carroll, K.S. and Herschlag, D. (2003) Annu. Rev. Biochem. 72, 517-571. “Challenges in Enzyme Mechanism and Energetics.” (Medline) (PDF File)
- Kraut, D.A., Sigala, P.A., Pybus, B., Liu, C.W., Ringe, D., Petsko, G.A. and Herschlag, D. (2006) PloS Biology 4e99 “Testing Electrostatic Complementarity in Enzyme Catalysis: Hydrogen Bonding in the Ketosteroid Isomerase Oxyanion Hole.” (Medline) (PDF File)
-
Sigala, P.A., Fafarman, A.T., Bogard, P.E., Boxer, S.G. and Herschlag, D. (2007) J. Am. Chem. Soc. 129, 12104-12105. “Do Ligand Binding and Solvent Exclusion Alter the Electrostatic Character within the Oxyanion Hole of an Enzymatic Active Site?” (Medline) (PDF File)
See also: Faculty of 1000 Biology, 16 Oct. 2007 http://www.f1000biology.com/article/id/1091226/evaluation -
Sigala, P., Kraut, D., Caaveiro, J., Pybus, B., Ruben, E., Ringe, D., Petsko, G., Herschlag, D., (2008) J. Am. Chem. Soc. 130, 13696-13708. “Testing Geometrical Discrimination within an Enzyme Active Site: Constrained Hydrogen Bonding in the Ketosteroid Isomerase Oxyanion Hole.”(Medline) (PDF File)
See also: Nature 2008, 456, 45-47. http://www.nature.com/nature/journal/v456/n7218/full/456045a.html
Enzyme Promiscuity, Evolution, and Phosphoryl Transfer
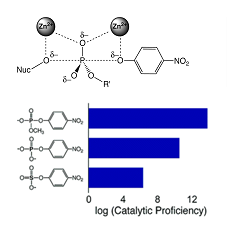 In recent years it has been recognized that proteins in the same superfamily –i.e.,
evolutionarily related proteins that possess similar structural motifs– often have low levels of activity toward reactions catalyzed by other members within the superfamily. This property, coined 'catalytic promiscuity', provides a powerful tool for addressing both evolutionary and mechanistic questions. We are exploiting this property of enzyme superfamilies to gain an understanding of the fundamental underpinnings of catalysis. Unlike traditional site-directed mutagenesis experiments that are often limited to studying a single reaction catalyzed by an individual enzyme, we are using a comparative approach to ask not simply what the consequence is of removing a particular side chain, but how that removal and change in the side chain affects normal and promiscuous reactions. Conversely, we can also ask how changes in an analogous residue in another member of the superfamily affect its cognate and promiscuous activity. Because the substrates varying in their charge, geometry, and transition state natures, the observed effects tell us more precisely about the catalytic roles of particular interactions and how these enzymes have become optimized over time for catalysis of their cognate reactions.
In recent years it has been recognized that proteins in the same superfamily –i.e.,
evolutionarily related proteins that possess similar structural motifs– often have low levels of activity toward reactions catalyzed by other members within the superfamily. This property, coined 'catalytic promiscuity', provides a powerful tool for addressing both evolutionary and mechanistic questions. We are exploiting this property of enzyme superfamilies to gain an understanding of the fundamental underpinnings of catalysis. Unlike traditional site-directed mutagenesis experiments that are often limited to studying a single reaction catalyzed by an individual enzyme, we are using a comparative approach to ask not simply what the consequence is of removing a particular side chain, but how that removal and change in the side chain affects normal and promiscuous reactions. Conversely, we can also ask how changes in an analogous residue in another member of the superfamily affect its cognate and promiscuous activity. Because the substrates varying in their charge, geometry, and transition state natures, the observed effects tell us more precisely about the catalytic roles of particular interactions and how these enzymes have become optimized over time for catalysis of their cognate reactions.
The Alkaline Phosphatase (AP) superfamily provides a powerful system in which to address these questions and also allows us to address the important and widespread biological reactions that involve phosphoryl transfer. Members of the AP superfamily catalyze a range of reactions including phosphoryl and sulfuryl transfer reactions. We currently focus work on studying two members of this superfamily, alkaline phosphatase (AP) and nucleotide pyrophosphatase/phosphodiesterase (NPP). AP preferentially catalyzes phosphate monoester hydrolysis reactions, whereas NPP is a proficient diesterase. Both enzymes have promiscuous activities toward each other's reactions. Although these enzymes catalyze different reactions with a specificity difference of >1013-fold, AP and NPP share an indistinguishable Zn2+ bimetallo site. We use a variety of structural and functional probes to elucidate the origins of this specificity difference. These approaches include steady stateand pre-steady state kinetic comparisons of cognate and non-cognate substrate reactions with wild type and mutant enzymes to determine rate enhancements and to dissect catalysis; structural comparisons between homologous enzymes to guide and interpret site-directed mutagenesis; X-ray crystallography and EXAFS to compare structures of homologous enzymes and to determine the structural consequences of mutations; vibrational spectroscopy to assess the nature of bound ligands; binding of substrates, inhibitors, and transition state analogs to wild type and mutant enzymes to further compare homologous enzymes and determine the energetic consequences of mutations; and linear free energy relationships and heavy atom isotope effects to obtain information about the reactions' transition states and their active site interactions.
Some leading papers from the lab in the areas of catalytic promiscuity and phoshoryl transfer are:
- O'Brien, P. and Herschlag, D. (1999) Chemistry and Biology 6, R91-R105. "Catalytic Promiscuity and the Evolution of New Enzymatic Activities." (Medline) (PDF File)
- Admiraal, S.J., Meyer, P., Schneider, B., Deville-Conne, D., Janin, J. and Herschlag, D. (2001) Biochemistry 40, 403-413. "Chemical Rescue of Phosphoryl Transfer in a Cavity Mutant of Nucleoside Diphosphate Kinase: Implications for Site-Directed Mutagenesis." (Medline) (PDF File)
- O'Brien, P. and Herschlag, D. (2002) Biochemistry 41, 3207-3225. "Alkaline Phosphatase Revisited: The Hydrolysis of Alkyl Phosphates." (Medline) (PDF File)
- Cheng, H., Nikolic-Hughes, I., Wang, J.H., Deng, H., O'Brien, P.J., Wu, L., Zhang, Z.Y., Herschlag, D. and Callender, R. (2002) J. Am. Chem. Soc. 124, 11295-11306. "Environmental Effects on Phosphoryl Group Bonding Probed by Vibrational Spectroscopy: Implications for Understanding Phosphoryl Transfer and Enzymatic Catalysis." (Medline) (PDF File)
- Zalatan, J.G., Fenn, T.D., Brunger, A.T., and Herschlag, D. (2006) Biochemistry 45, 9788-9803 "Structural and Functional Comparisons of Nucleotide Pyrophosphatase/Phosphodiesterase and Alkaline Phosphatase: Implications for Mechanism and Evolution."(Medline) (PDF File)
- Zalatan, J., Fenn, T. D., Herschlag, D. (2008) J. Mol. Biol. 384, 1174-1189. "Comparative Enzymology the Alkaline Phosphatase Superfamily to Determine the Catalytic Role of an Active Site Metal Ion." (Medline) (PDF File)