Equicksearch
- EQUICKSEARCH*
EQUICKSEARCH*
FUNCTION
EQuickSearch rapidly identifies places where query sequence(s) occur in a nucleotide sequence database. The output is a file of overlaps that can be displayed with QuickMatch or EQuickShow. You can make up your own sequence database or use GenEMBL, which consists of GenBank and those sequences in EMBL that are not represented in GenBank (or the other way around at some sites).
NOTE: The EGCG Quick Searching System programs are now fully supported by the EGCG team. GCG distributed the original programs in the hope that users would make suggestions about their future development. This program is one such suggestion.
DESCRIPTION
EQuickSearch answers the question, "Does this sequence occur in a sequence database?" It is not designed to look for distantly related sequences, but can be very useful (and very quick) in finding duplicates or identifying minor differences.
The search can be set up to be sensitive enough to find all primate beta globin genes when a human beta globin is used as the probe. The search is robust enough to tolerate up to two errors per 100 base pairs, in queries of 300 base pairs or longer. These errors can be either substitutions or insertion/deletion errors (gaps). The search finds the query sequence in the database on either strand in any circular permutation.
The output is a list of the places in the database (overlaps) where some number (stringency) of words from your query match within some number (window) of possible words. The list may contain a few spurious overlaps or false positives.
The best way to look at the list is to display the overlaps with the QuickMatch program. QuickMatch makes optimal alignments of the regions of overlap. You can exclude alignments that fail to meet stringency criteria, and exclue self matches. You can also limit the output to just the list of accepted hits and display only the mismatches rather than the complete alignments. QuickMatch can also make a dot-plot of each region where an overlap occurred. The dot-plots let you visualize the exact relationship between the query and any sequence it overlaps in the database. If you do not have a fast graphics device,
AUTHOR
This GCG program was modified by Peter Rice (E-mail: pmr@sanger.ac.uk Post: Informatics Division, The Sanger Centre, Hinxton Hall, Cambridge, CB10 1RQ, UK).
All EGCG programs are supported by the EGCG Support Team, who can be contacted by E-mail (egcg@embnet.org).
EXAMPLE
Here is a session with EQuickSearch that was used to find sequences in the EMBL data library with similarities to the coding sequence for the human fetal beta globin G-Gamma.
% equicksearch
EQUICKSEARCH uses nucleotide sequences
EQUICKSEARCH of what sequence(s) ? ggammacod.qeq
Search for query in what hash table:
1 emnew1 em_new
2 emnew2 em_new
3 genembl1 em_ba, em_est
4 genembl2 em_est
5 genembl3 em_est, em_fun
6 genembl4 em_fun, em_in, em_om, em_or
7 genembl5 em_or, em_ov, em_pat, em_ph, em_pl, em_pr
8 genembl6 em_pr, em_ro
9 genembl7 em_ro, em_sts, em_sy, em_un
10 genembl8 em_un, em_vi
Select number, range or name (* all *) ?
What is the window size (* 15 *) ?
What is the stringency (matching words per window) (* 7 *) ?
What should I call the output file (* ggammacod.quick *) ?
Reading EQuickDir:emnew1 . . . . .
Ggammacod Sgmt: 1 Strd "+" Len: 444...... Overlaps: 1
Ggammacod Sgmt: 1 Strd "-" Len: 444...... Overlaps: 0
/////////////////////////////////////////////////
Reading EQuickDir:genembl8 . . . . .
Ggammacod Sgmt: 1 Strd "+" Len: 444...... Overlaps: 0
Ggammacod Sgmt: 1 Strd "-" Len: 444...... Overlaps: 0
Overlaps found with this group of 1 sequences: 17
EQuickSearch with what query sequence(s) ?
Queries: 1
Total Length: 444
Overlaps: 17
CPU (sec): 23.8
Output file: ggammacod.quick
%
OUTPUT
EQuickSearch makes an output file with a list of the overlaps between your query sequence and the database. The alignments and dot-plots from this example session are shown with QuickMatch. Here is the output file from this session:
! EQUICKSEARCH of: Ggammacod.Seq February 1, 1989 18:41 ! WordSize: 20 Window: 15 Stringency: 7 .. \\ D22:[Burgess.Work]Ggammacod.Seq;1 Sgmt: 1 Strd: + ! Coding sequence for Human fetal beta globin G-gamma. !Sequence Sgmnt Words Documentation Primate:Chpagglog 1 18 !Chimpanzee fetal A-gamma-globin gene. Primate:Chpggglog 1 17 !Chimpanzee fetal G-gamma-globin gene. Primate:Humhbb 9 21 !Human beta globin region on chromosom Primate:Humhbb 10 14 !Human beta globin region on chromosome Primate:Humhbgab 1 16 !Human A-gamma-globin gene on chromosome Primate:Humhbgg 1 21 !Human glycine-gamma-globin, 3' end. Primate:Machbca2 1 17 !Rhesus monkey beta-cluster 5' fetal Primate:Machbga1 1 15 !Rhesus monkey beta-cluster 3' fetal Primate:Orahbg1f 1 19 !Orangutan gamma-1-fetal globin gene Primate:Orahbg2f 1 17 !Orangutan gamma-2-fetal globin gene Embl:Ggagglog 1 18 !Gorilla fetal A-gamma-globin gene. Embl:Ggggglog 1 20 !Gorilla fetal G-gamma-globin gene. Embl:Hsggl2 1 19 !Human a gamma-globin gene. 3/83 Embl:Hsggl3 1 16 !Human A-gamma-globin gene. 8/83 Embl:Hsggl4 1 21 !Human G-gamma-globin gene. 12/83 Embl:Hsglbn 1 20 !Human gene for fetal A-gamma and Embl:Hsglbn 2 14 !Human gene for fetal A-gamma and //
RELATED PROGRAMS
QuickMatch displays the overlaps found by EQuickSearch with either optimal alignments or dot-plots. The alignments can be selected by quality to discard poor matches. The dot-plots can be reviewed rapidly with a graphic screen. EQuickIndex builds hash tables from sequence(s) in data libraries, and stores them as map sections. These tables make up the database that is searched by EQuickSearch
DataSet creates a GCG data library from any set of sequences in GCG format.
WordSearch identifies sequences similar to a query sequence using a Wilbur and Lipman search. WordSearch answers the question, "What sequences in the database are similar to my sequence?" The output is a list of significant diagonals whose alignments can be displayed with Segments. ProfileSearch uses a profile (representing a group of aligned sequences) as a query to search the database for new sequences with similarity to the group. The profile is created with the program ProfileMake.
TWordSearch identifies DNA sequences similar to a protein query sequence using a six frame translation of the database and a Wilbur and Lipman-style search. The output is a list of significant diagonals whose alignments can be displayed with TSegments. TProfileSearch uses a profile (representing a group of aligned protein sequences) as a probe to search the nucleotide database for new sequences with possible protein products having some similarity to the group. The profile is created with the program ProfileMake.
RESTRICTIONS
If the query sequence is longer than 4,200 base pairs, it is divided into segments of 4,200 base pairs and each segment is searched as if it were a different sequence. Each segment overlaps the one before it by 200 base pairs (10 word lengths).
SELECTING TABLES
You use the tool EQuickIndex to make up your own hash tables from GCG data libraries. This topic tells you how to make your tables known to EQuickSearch so that you can select a table that you have created.
EQuickSearch reads a local data file called equicksearch.menu in directory EGenRunData: to find out what hash tables are available for searching. If you construct your own hash tables with EQuickIndex, you will want to have your own copy of equicksearch.menu in your own directory, or use the command line option -INDEXmenu=filename to specify a file with the names of the tables that interest you.
If EQuickSearch reads a menu file with more than one table, it will present you with a menu and you can select the table that interests you. The menu and your response could look something like this:
Search for query in what hash table:
1 emnew1 em_new
2 emnew2 em_new
3 genembl1 em_ba, em_est
4 genembl2 em_est
5 genembl3 em_est, em_fun
6 genembl4 em_fun, em_in, em_om, em_or
7 genembl5 em_or, em_ov, em_pat, em_ph, em_pl, em_pr
8 genembl6 em_pr, em_ro
9 genembl7 em_ro, em_sts, em_sy, em_un
10 genembl8 em_un, em_vi
Please type one name completely (* all *): genembl
In reply,
you can either specify a set of index names,
for example genembl*,
or you can specify the database division the index was derived from using any substring (such as ?em_est to get every index that includes the EST division).
You can suppress this menu by putting your choice on the command line with an expression like: -TABle=genembl.
Although each hash table is made up of four different files, all of these share a common name and directory. The table you choose is the file name for the hash table. A file of the name you have chosen must be present in the list of possible hash tables in equicksearch.menu in directory EGenRunData:.
SUGGESTIONS
Increasing Sensitivity
If you think there is a sequence like your query in the database but you don't find it, you can increase the sensitivity of the search. If your query is different from the sequence in the database by as much as 2 differences per 100 base pairs, you will have to try lowering the stringency or increasing the window size or both. Try a window of 20 and a stringency of 5 to start off.
False Positives
The default settings for window and stringency of 15 and 7 should give very few false positives, but this may be more stringent than you really want. If you set the stringency down or the window up, there are more opportunities for divergent sequences to match, but likewise there are more opportunities to find spurious overlaps. Whenever more than one spurious overlap is expected, the expectation is printed beneath each query segment in the output file.
Decreasing Sensitivity
There are times when you only want to find exact matches of a query in the database. EQuickSearch provides two switches to help you restrict the overlaps found to exact matches. -ONEstrand restricts the search to the top strand of the query only. -PERfect lets EQuickSearch set the window and stringency as high as possible for each query, depending on its length. Using this switch, EQuickSearch will set the window to the number of possible words in your sequence and the stringency to three less than the window. EQuickSearch cannot set the window higher than 50, but a requirement of 47 matching words in 50 is VERY STRINGENT. Do not use -PERfect if there are ambiguity codes in the query.
If you want to find perfect overlaps with other sequences which may be shorter than the full length of your search sequence, you should instead use less stringent swindow and stringency values, and use QuickMatch- Perfect to check the matches reported.
Identifying the Query Set
See the Data section of the User's Guide and Appendix VI for information about naming groups of sequences.
CONSIDERATIONS
The Match Criterion is Very Stringent
The criterion for a word in your query to find a corresponding word in the database is that a particular group of 20 of the 40 possible bits in their 2-bit codes match exactly. There is no symbol comparison table. Lower and upper case letters are equivalent however.
False Positives
The default settings for window and stringency of 15 and 7 should generate very few false overlaps if your sequence does not contain generic sequence elements. The best way to recognize false positives is to filter the output with QuickMatch. You should remember that two 20-mers can hash to the same bucket without having a single base in common!
QuickMatch is able to tolerate a large number of false positives by screening the quality of each alignment before writing it. You can set the stringency low (or the window number high) for EQuickSearch and use the EQuickSearch -STRINgency=0.5 to eliminate most of the false positives.
Repetitive Sequences and Simple Sequences Confound EQuickSearch
If you use a query sequence that contains a mammalian Alu-family sequence, you are in danger of finding the other Alu-family sequences. Polymers like (TAA)(n), (TG)(n), and polyA will overlap with all the places in the database where similar polymers occur. Worse, simple repeats will overlap several times and give very high scores.
Short Query Sequences
You can use short queries if you set the window down, but you should realize that EQuickSearch was designed to look for sequences longer than 300 base pairs. You should set the window size to be less than 1/20th of the length of your sequence.
Of course, if your query sequence is too short (less than 40 bases) there may be no matching word indexed in the database.
Short Sequences in GenEMBL
Some sequences in the database may be too short to have enough words indexed. The stringency is the minimum number of words that EQuickSearch must find to identify a match, so short sequences will be ignored. The minimum length that you can find will be 20 times the stringency you specify plus a few bases. For the default value of 7, this is a minimum length of about 160 bases.
Ambiguity Codes
The corruption of the 20 bits in the hash mask by ambiguity codes is subtle (see algorithm below). Since each base informs only one bit in the mask, there is at least a 50 percent chance that any particular ambiguity code will inform its bit correctly. With codes like R and Y, one of the bits can be assigned correctly in every event and the other bit is random. EQuickSearch and EQuickIndex (the utility that generates the hash table from the database) both ignore all words that have more than two ambiguous bits in them. To a first approximation, you can assume that any word in the database or the query with more than two ambiguity codes will be ignored.
Errors in the Query
The EQuickSearch algorithm is designed to find any sequence in the database longer than 76 base pairs when it is searched with an identical probe. Since DNA cannot be determined to 100 percent accuracy, the algorithm has been designed to be robust to small numbers of errors in the query. There is a discussion of errors in Devereux (submitted for publication to CABIOS, preprints available from GCG).
In an experiment to validate this algorithm, we took sequence fragments from GenEMBL that were 300 base pairs long. We randomly introduced three substitution and three short length errors (less than four base pairs) into these sequence fragments. We found that 95 percent of these corrupted sequences could still find their original versions in GenEMBL. Nonetheless, there is a small number of permutations of 6 errors in 300 base pairs that will make the default settings of window and stringency miss the sequence of interest. If you suspect that your sequence has a lot of errors, you will have to reduce the stringency or increase the window or both.
ALGORITHM
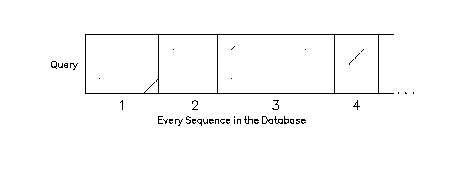
EQuickSearch uses the algorithm of Devereux (submitted for publication to CABIOS) to compare sequences ("queries") to a sequence database. The database is represented with a sparse hash table. Visualize the comparisons as a set of dot-plots with the query as the vertical sequence and the sequences in the database to which the query is being compared as the different horizontal sequences. The search finds contiguous regions of sequences in the database that share more than some set number of words with your query.
Hash Codes

Every possible word of length 20 can be categorized with a hash function. The function we use divides 20-mers up into 1,048,576 different categories. First, the sequence is translated into binary using G=11, A=10, T=01, and C=00. Then a mask is used to cover 20 of the 40 bits in the 20-mer's binary representation. Last, the 20 bits left uncovered by the mask are used to calculate an index or "hash code" between 0 and 1,048,575.
Making a Hash Table
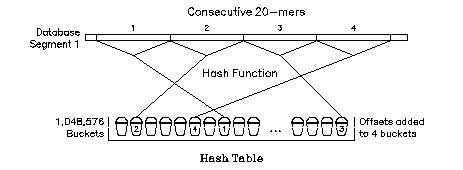
Before the search, each adjacent word in the database of length 20 is assigned a discrete offset from the beginning of the database. The offset is the distance of the word from the beginning of the database in units of word length. A hash code for each of the 20-mers is calculated as described above. The offset for each word is then put into a "bucket" with the other words that share its hash code. When all the adjacent words have been divided up and sorted into their respective buckets, the contents of all the buckets are stored on disk. These buckets are collectively referred to as a "hash table." Notice that 19/20 of the words in the database are not put into the hash table since only the offsets from adjacent words are kept!
Searching in the Hash Table
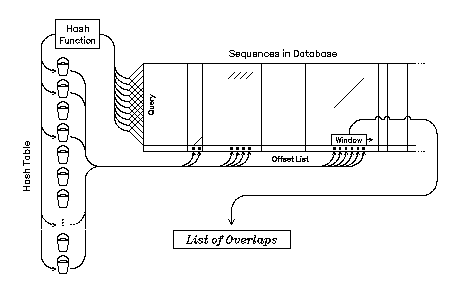
When you present a query to EQuickSearch it looks in the buckets that correspond to every 20-mer in your query sequence. The offsets found in these buckets tell where words with the same hash patterns as those in your query occur in the database. The offsets from each bucket are sorted into an offset list. A window of set width slides along the whole offset list looking for places where some set threshold (stringency) of offsets occurs. If stringency or more offsets occur at any position of the window, a record is added to the output file that shows where this overlap occurs and the window jumps to the next segment of the offset list.
The Output List
The output is a list of the places in the database where some number of words (stringency) occur within some range of possible words (window).
The Segment Concept
If the query sequence is longer than 4,200 base pairs, it is divided into segments and each segment is searched as if it were a different query sequence. Each segment overlaps the one before it by 200 base pairs (10 word lengths). Although 15 is the default window setting, you will find some redundant overlaps at the point where query segments overlap.
Any sequence in the database found by EQuickSearch is also assigned a segment number. Ninety-five percent of the sequence entries in GenEMBL are shorter than 4,200 base pairs. The segment number for all of these is simply one. For longer sequences, the segment number tells you where in the database sequence (within 4,200 base pairs) the overlap occurred. QuickMatch likewise displays one segment of overlap at a time.
Words
For each segment in the database where an overlap occurs, the output shows how many words were in common between the query and that particular segment.
COMMAND-LINE SUMMARY
All parameters for this program may be put on the command line. Use the option -CHEck to see the summary below and to have a chance to add things to the command line before the program executes. In the summary below, the capitalized letters in the qualifier names are the letters that you must type in order to use the parameter. Square brackets ([ and ]) enclose qualifiers or parameter values that are optional. For more information, see "Using Program Parameters" in Chapter 3, Basic Concepts: Using Programs in the GCG User's Guide.
Minimum Syntax: % equicksearch [-INfile1=]ggammacod.seq -Default Prompted parameters: -TABle=?est the database(s) to be searched (default all) -WINdow=15 the window -STRingency=7 the stringency [-OUTfile=]ggammacod.quick the output file name Local Data Files: -INDEXmenu=equicksearch.menu list of tables available for searching Optional Switches: -MINLen=56 ignores any query less than 56 bp long -PERfect minimizes the number of overlaps found -ONEstrand searches only the top strand of your query sequence -NOMONitor suppresses the screen trace showing each query -NOSUMmary suppresses the summary at the end of the session
ACKNOWLEDGEMENT
GCG's original QuickSearch was developed by John Devereux of GCG in collaboration with Chemical Abstracts Service (CAS). The research was originally described in a Ph.D. thesis entitled "A Rapid Method for Identifying Sequences in Large Nucleotide Sequence Databases" (1988, reprints available from GCG).
The EGCG version of EQuickSearch was written by Peter Rice while working at EMBL, Heidelberg, Germany. It is based on the original version, with mapped sections used as index files on VMS and with large buffers to read the indices on Unix. The default search is redefined as all available database indices. The early version at EMBL, used by the original QUICK E-mail service, gad six databases defined for Quick Searching according to the sizes of the EMBL database divisions. The revised EQuickSearch uses indices split simply split by size, and allows the user to select a single database division if required.
Although GCG have dropped the original QuickSearch, we have found the modified EQuickSearch to be extremely valuable in a number of cases. At the Sanger Centre, Kate Rice has used it to compare a set of sequences to themselves by indexing her own customized database. The complete search takes only a few minutes for several hundred sequences. Peter Rice has similarly used the EGCG programs to find all overlaps of Escherichia coli sequences in the EMBL database. We are not aware of other programs capable of performing these tasks.
LOCAL DATA FILES
The files described below supply auxiliary data to this program. The program automatically reads them from a public data directory unless you either 1) have a data file with exactly the same name in your current working directory; or 2) name a file on the command line with an expression like -DATa1=myfile.dat. For more information see Chapter 4, Using Data Files in the User's Guide.
The file EGenRunData:equicksearch.menu contains the names of the hash tables available for searching. This file is automatically updated by EQuickIndex when new index files are generated. See the topic SELECTING TABLES above.
OPTIONAL PARAMETERS
The parameters and switches listed below can be set from the command line. For more information, see "Using Program Parameters" in Chapter 3, Basic Concepts: Using Programs in the GCG User's Guide.
-PERfect
sets the window and stringency dynamically as a function of the length of each query sequence. This option minimizes the number of overlaps that are not identical to the query (false positives).
-ONEstrand
searches only the top strand of your query sequence.
-MINLen=56
makes EQuickSearch ignore query sequences shorter than 56 base pairs.
-MONitor
This program normally monitors its progress on your screen. However, when you use the -Default option to suppress all program interaction, you also suppress the monitor. You can turn it back on with this option. If your program is running in batch, the monitor will appear in the log file. If the monitor is slowing the program down, suppress it with -NOMONitor.
-SUMmary
writes a summary of the program's work to the screen when you've used the -Default qualifier to suppress all program interaction. A summary typically displays at the end of a program run interactively. You can suppress the summary for a program run interactively with -NOSUMmary.
Use this qualifier also to include a summary of the program's work in the log file for a program run in batch.
REFERENCES
Devereux, J. (1989). A Rapid Method for Identifying Sequences in Large Nucleotide Sequence Databases. In preparation for submission to Computer Applications in the Biosciences. (This paper is based on a Ph.D. thesis of the same name, available from the Genetics Computer Group, Inc., University Research Park, 575 Science Drive, Suite B, Madison, Wisconsin, USA, 53711.)
Printed: April 22, 1996 15:53 (1162)