Twordsearch
- TWORDSEARCH(+)
TWORDSEARCH(+)
FUNCTION
TWordSearch identifies DNA sequences similar to a protein query sequence using a six frame translation of the database and a Wilbur and Lipman-style search. The output is a list of significant diagonals whose alignments can be displayed with TSegments.
DESCRIPTION
TWordSearch uses an algorithm similar to the algorithm of Wilbur and Lipman (Proc. Natl. Acad. Sci. (USA) 80; 726-730 (1983)) to compare one sequence (the query) to any group of sequences. You should think of the comparisons as a set of dot-plots with the query as the vertical sequence and the group of sequences to which the query is being compared as the different horizontal sequences (the search set). The search finds the registers of comparison (diagonals) that have the largest number of short perfect matches (words). The best segment of similarity along each diagonal can be viewed with the program TSegments.
What is a Word
A word is any short sequence (n-mer) where you have set n to some small constant like six or seven. The word 'GGATGGC' is one of the 16,384 possible words of length 7 that can be created from an alphabet consisting of the four letters G, A, T, and C. The word QQL is one of the 8,000 possible words of length 3 that you can make with the 20 letters of the amino acid alphabet.
What is a Word Mask
The symbols that match between two words need not be contiguous. You could use a '+' and a '-' to define a word mask like '++-++-++'. This mask would mean that matching words should match at positions 1, 2, 4, 5, 7, and 8 and that positions 3 and 6 may or may not match.
What is a Diagonal
A diagonal is a register of comparison for two sequences -- a path across a surface of comparison where X - Y for every point is a constant. A series of dots along a diagonal represent a segment of similarity between two sequences. Each diagonal can be defined by the constant X - Y for that diagonal. The path up from the origin is numbered zero. The paths above the zero diagonal are negative and the paths below the zero diagonal are positive. The diagonals are then numbered between minus the length of the vertical (query) sequence and plus the length of the horizontal (search set) sequence.
What is the Output
TWordSearch sorts the scores of all the diagonals in your comparison and shows you a list of the best diagonals where you have restricted the size of the list to some finite number like 50 or 100. You can see optimal alignments of the segments of similarity in the TWordSearch output file with the TSegments program.
List File
TWordSearch compares both strands of your query sequence to any set of sequences you name and shows the best diagonals and the number of symbols within matching words on each of these best diagonals. The diagonals are identified with the coordinate X - Y (described above), the number of symbols within the matching words for that diagonal, the strand of the query sequence, and the name of the search set sequence.
Score Distribution Plot
TWordSearch makes a histogram showing the number of diagonals observed for each diagonal score. The histogram shows the distribution of diagonal scores so you can see if a particular diagonal in your list of best diagonals is significant.
AUTHOR
This GCG program was modified by Peter Rice (E-mail: pmr@sanger.ac.uk Post: Informatics Division, The Sanger Centre, Hinxton Hall, Cambridge, CB10 1RQ, UK).
All EGCG programs are supported by the EGCG Support Team, who can be contacted by E-mail (egcg@embnet.org).
EXAMPLE
Here is a session using TWordSearch to find sequences in the GenEMBL nucleotide sequence data library with similarities to a human globin coding sequence.
% twordsearch -PLOt
TWORDSEARCH uses protein sequence data
TWORDSEARCH of what sequence ? Sw:laci_ecoli
Start (* 1 *) ?
End (* 360 *) ?
TWORDSEARCH uses nucleotide sequences
What sequence(s) ? GenEMBL:ec*
What word size (* 2 *) ?
List how many best diagonals (* 50 *) ?
Integrate how many adjacent diagonals (* 3 *) ?
What should I call the output file (* laci_ecoli.word *) ?
1 ec01911 Len: 2,139
101 ecc625 Len: 96
/////////////////////////////////////
Output file: laci_ecoli.word
When your LaserWriter attached to tty07 is ready, press .
%
OUTPUT
TWordSearch makes an output file of sequence names with a list of the best diagonals in your search and plots the distribution of scores from the search. Here is some of the output file:
(Peptide) WORDSEARCH of: Sw:Laci_Ecoli check: 1939 from: 1 to: 360 ID LACI_ECOLI STANDARD; PRT; 360 AA. AC P03023; DT 21-JUL-1986 (REL. 01, CREATED) DT 21-JUL-1986 (REL. 01, LAST SEQUENCE UPDATE) DT 01-AUG-1991 (REL. 19, LAST ANNOTATION UPDATE) DE LACTOSE OPERON REPRESSOR. . . . TO: GenEMBL:Ec* Sequences: 3,041 Total-length: 5,339,505 September 7, 1993 12:02 Word-size: 2 Words: 39357944 Diagonals: 4,477,569 Total-diagonals: 6,431,224 Integral-width: 3 Alphabet: 20 List-size: 50 CPU minutes: 5.94 Sequence Strd Diag Score Width Documentation .. Empro:Eclaci + 10 362 3 V00294 E. coli laci ... Empro:Eclac + 26 362 3 J01636 E.coli lacto ... Empro:Eclact41 + 0 361 3 X58469 E.coli T41 m ... /////////////////////////////////////////////////////////////// Empro:Ecfhla + 1199 31 8 M58504 E.coli forma ... Empro:Ecfepcdg + -9 31 13 X57471 E.coli fepC, ... Empro:Ecfecbcde + 3334 31 11 M26397 K-12 fecA ge ...
SCORE DISTRIBUTION PLOT
If you run TWordSearch with the command line option -PLOt, it plots a histogram showing the number of diagonals observed with each different score. This plot should help you judge which of the diagonals in your output list are significant and whether the output list was large enough to contain all of the significant diagonals.
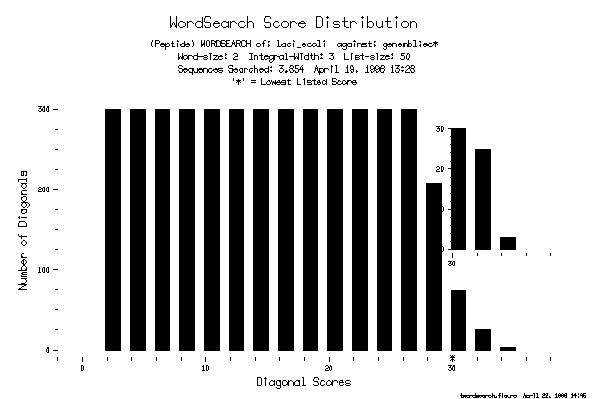
This is the plot from the example.
Bin Size
You can set the resolution of the score distribution plot with a command line expression like -BINsize=6. By default, each histogram is integrated into bins that are the size of the word length. For words of length 6, the histograms would normally show the frequency of diagonals with scores from 0 to 5, 6 to 11, 12 to 17, et cetera.
The Histogram Shows Scores for Structures
The histogram shows the scores for diagonals after processing into structures. See the ALGORITHM topic below for a description of how scores accumulate on diagonals and the way scores are grouped into structures before becoming eligible to join the list of best diagonals.
List Cutoff
Ideally the list of best diagonals should be large enough to include some diagonals from the high end of the random scores. The list of best diagonals may not have been large enough, however, to show all of the diagonals with significant scores. The cutoff or lowest score in the output list is marked on the "Diagonal Scores" axis with an asterisk (*). Notice that the list size was not large enough to include all of the globin sequences in GenEMBL.
Blowup
The end of the histogram with the best observations (highest scores) is magnified into a small plot in the upper-right corner. The inset plot simply expands the vertical axis tenfold so that the number of high-scoring diagonals can be read exactly.
RELATED PROGRAMS
TSegments aligns and displays the segments of similarity found by TWordSearch WordSearch identifies sequences similar to a query sequence using a Wilbur and Lipman search. WordSearch answers the question, "What sequences in the database are similar to my sequence?" The output is a list of significant diagonals whose alignments can be displayed with Segments. Segments aligns and displays the segments of similarity found by WordSearch.
If you run Compare with the command line option -WORd, it calculates the points for a a dot-plot that shows where common words between two sequences occur.
EQuickSearch rapidly identifies places where query sequence(s) occur in a nucleotide sequence database. The output is a file of overlaps that can be displayed with QuickMatch or EQuickShow. You can make up your own sequence database or use GenEMBL, which consists of GenBank and those sequences in EMBL that are not represented in GenBank (or the other way around at some sites). ProfileSearch uses a profile (representing a group of aligned sequences) as a query to search the database for new sequences with similarity to the group. The profile is created with the program ProfileMake. TProfileSearch uses a profile (representing a group of aligned protein sequences) as a probe to search the nucleotide database for new sequences with possible protein products having some similarity to the group. The profile is created with the program ProfileMake. FastA does a Pearson and Lipman search for similarity between a query sequence and any group of sequences. For nucleotide database searches, FastA is more sensitive than BLAST. TFastA does a Pearson and Lipman search for similarity between a query peptide sequence and any group of nucleotide sequences. TFastA translates the nucleotide sequences in all six reading frames before performing the comparison. It is designed to answer the question, "What implied peptide sequences in a nucleotide sequence database are similar to my peptide sequence?"
FindPatterns, StringSearch and Names are other sequence identification programs.
RESTRICTIONS
The query sequence may not be more than 30,000 symbols long. You may not select a list size of more than 1,000 "best" diagonals. The word size should be from 1 to 30. Word searching is subject to many limitations and considerations, which are discussed further below.
CONSIDERATIONS
The Match Criterion
The match criterion for two words is that all of the symbols in each word are identical. The symbols that must be identical need not be contiguous if a word mask has been set, but the symbols that must match must be identical, except for case. There is no symbol comparison table and no support for the equivalence of nucleic acid ambiguity codes. Lower and upper case letters are equivalent however.
Word Searching Requires some Perfect Identity
The basic assumption of word comparisons is that patterns of similarity have an unusual number of common words (short perfect matches) along a set of closely spaced diagonals. This is often the case for nucleic acid sequences that have diverged recently, but it may not be true for peptide comparisons. You should consider this assumption carefully. When two sequences have diverged sufficiently so that an optimal alignment of them has one mismatch for every six bases, then a word comparison with words of length 6 may not recognize their similarity.
Sequence Simplification May Increase the Level of Perfect Identity
The command line option -SIMplify allows you to map the sequences' symbols into a simpler subset of symbols to find matches between categories of sequence symbols.
Queries Containing Repetitive or Simple Sequences
If you use a query sequence containing a mammalian Alu-family sequence, you are in danger of finding the hundreds of Alu-family sequences that have been published to the exclusion of everything else. The ideal query sequence contains no simple (e.g., =polyA) or repeated sequences. Ideally the query should be short enough so that any segment of similarity generates an unusual peak on the histogram. If the query is less than 500-bases long, most of the diagonals are approximately the same length. Short diagonals of similar length increase the probability that word scores from a small segment of similarity are not lost in the background noise.
SUGGESTIONS
Word Size
You might try a word size of 6 and an integral width of 3 for nucleic acid searches as suggested by the program's defaults. You should recognize that when the average word occurs in the query sequence more than zero times, the amount of cpu time rises dramatically. Notice in the example above that fewer than 10% of the diagonals contained any common words of length 6. You could start with a word size of 2 for peptide sequence comparisons.
Word Mask
A word mask calling for two matches followed by one uncertainty is more sensitive for recognizing protein coding sequences than a simple contiguous word search. You can set up a word mask by including an expression like -MASk=++-++-++ on the command line, using a '+' (plus sign) to show the positions where the symbols must match and a '-' (minus sign) to show the positions where symbols may or may not match). Wobble in the third codon position in the genetic code would make a mask like '++-++' more sensitive than '+++++' for recognizing similar coding regions.
It does not make sense to define a mask with leading or trailing '-' characters, and therefore TWordSearch removes these. Defining a word mask suppresses the word size query since the word size inheres to the mask you have chosen. The word size of the mask '++-++-++' is 8, even though only six of the eight characters under the mask must match.
List Size
The list should be large enough to cover all of the significant scores with at least 10 scores seeming to arise from the high end of the random scores. The default list size of 50 is large enough for most query sequences, but it is not large enough to include all of the globins in the sample session.
Identifying the Search Set
See the Specifying Sequences section of the User's Guide for information about naming groups of sequences.
Batch Queue
TWordSearch is one of the few programs in the EGCG Program Library that can take more than a few minutes to run. Most comparisons should probably be run in the batch queue. You can run this program in the batch queue on many computers by using the command-line option -BATch. Run this way, the program prompts you for all the required parameters and then automatically submits itself to the batch or at queue. Batch jobs free your terminal for other work and may allow the system manager to distribute the load on your computer more evenly. For more information, see "Using the Batch Queue" in Chapter 3, Basic Concepts: Using Programs in the User's Guide. Very large comparisons may exceed the cpu limit set by some systems.
When TWordSearch is run in batch as % twordsearch -PLOt -BATch, instructions for plotting the optional histogram is written to a figure file named wordsearch.figure, unless the plot has been directed to a specific file or graphics device from the command line. Please see the Figure entry in the Program Manual for instructions on how to plot a figure file to any graphics device that GCG supports.
Short Peptide Query Sequences
TWordSearch tries to guess if your query is a protein or nucleic acid sequence. If the query sequence really is a protein, but TWordSearch guesses wrong, you will see the alphabet parameter set equal to 4 and the word "Nucleotide" next to "WordSearch" at the top of the output list. The search may not find any word matches at all! The command line option -PROtein tells TWordSearch to make the word search based on the whole alphabet of sequence characters found in the query.
Interrupting a Search: You can type
The database programs Names,
StringSearch,
FindPatterns,
FastA,
TFastA,
and WordSearch
can be used for list refinement if you are looking for sequences with something in common.
For instance,
you could identify human globin sequences with StringSearch.
The output list could then be refined with FindPatterns
to show only those globin sequences containing EcoRI sites.
You could then use WordSearch
to compare this output list to a sequence of your own that you think is similar to these human,
globin,
EcoRI-containing sequences.
You can add two lists together by simply appending one of the files to the other.
It is better if you use a text editor to modify the heading of the combined list so that the annotation in the list correctly reflects what you have done.
Remember to delete the text heading from the second file so that it does not occur in the middle of the list.
Suppress any item in a list by typing an exclamation point (!)
in front of the item.
You can also put comments into a list anywhere on a line by placing an exclamation point before the comment.
TWordSearch assembles a list of the best places in your search set to look for similarities to your query sequence.
The output is in the file of sequence names format and is therefore suitable for input to any program that allows indirect file specifications.
(See the Specifying Sequences
section of the User's Guide
for information about indirect file specification.)
The first part of the output file contains heading information about the parameters of the search,
including a definition of the query sequence,
the word size,
the window of integration,
the size of the desired list,
the number of symbols found within matching words (after integration),
the number of diagonals on which those words were found,
the total number of diagonals in the search,
and the size of the alphabet of symbols used.
Several lines of the TWordSearch output file must be readable to the TSegments
program.
The second part of the file contains the list of significant diagonals.
These diagonals are defined by the following features: the sequence name,
the strand ('+' or '-'),
the X - Y coordinate that identifies the peak diagonal (Diag),
the number of symbols on the diagonal that were within matching words (Score)
,
the width of the structure (Width),
and a short line of documentation.
All of this information is read by the TSegments
program.
(See the ALGORITHM
topic below for a further explanation of the information listed with each significant diagonal.)
The algorithm described below may be referred to as a hash-table/linked-list search.
Wilbur and Lipman searches are an example of a class of comparisons that use direct addressing or k-tuple preprocessing to reduce search time.
You set a word size or defines a word mask,
which implies a word size.
Then TWordSearch makes up a dictionary of all of the possible words of that size in the query sequence.
A second dictionary is compiled for the opposite strand if the query is a nucleic acid sequence.
The dictionary has an entry for every possible word.
Imagine each word,
such as 'GGATGG',
as a number in base 4 that corresponds to an entry in the dictionary.
At each entry,
there is a number telling the positions (coordinates)
where the word occurs in the query sequence.
If the word does not occur,
the number at the entry is zero.
Then,
for each word in the searched sequences,
TWordSearch just looks up the word in the dictionary to find out if it occurs in the query sequence.
If the word from a search set sequence does occur in the query sequence,
TWordSearch adds the length of the word to the score for the diagonal on which the word occurs.
If a word match overlaps another one,
only the new symbols are added to the score for the diagonal.
For instance,
two adjacent word matches of length 6 would contribute a total of seven to the score for their diagonal.
The parameter alphabet that appears in the output is the number of symbols that could make up each word.
For peptide sequences,
the alphabet is the number of sequence symbols that were actually used in the query sequence.
The alphabet should be four for nucleic acids.
Notice that nucleic acid ambiguity codes are not supported by this alphabet and that they confound word comparison! Every word in any search set sequence that contains characters that are not part of the comparison "alphabet" is ignored.
U and T are equivalent in nucleic acid sequences,
however,
so DNA patterns may be found in RNA sequences.
Upper case and lower case sequence symbols are equivalent in all comparisons.
The Histogram: Score
An array of counters,
one for the score on each diagonal,
is maintained.
Each time a word is found in both the horizontal and vertical sequences,
the counter for the diagonal on which it was found is incremented by the number of symbols in the word.
After each sequence is searched with the dictionary from the query sequence,
the result is an array of numbers that tells how many symbols occur within matching words along each diagonal of the comparison.
This array of diagonal counters is referred to as the histogram.
To make the search more tolerant of short length differences (gaps)
between the query and the sequences in the database to which it is similar,
TWordSearch combines the scores of a user-defined number of adjacent diagonals and puts the combined score (rounded up)
at the center of this "window of integration." Wilbur and Lipman call this region of adjacent diagonals a window-space.
Finding the N-Best Diagonals: Structures After integration,
the histogram is searched for a position in which there is a score above the average.
A structure is defined as a region of diagonal scores in the integrated histogram from the first above-average score to the last;
that is,
to where the scores fall back to the average again.
If the peak score for a structure is better than the worst score in the list of the N-best diagonals observed so far,
then the structure is put in the list and the existing worst observation in the list is discarded.
The structure is recorded by recording the file and entry being searched,
the coordinate of the diagonal at the center of the peak region rounded up,
the peak score (after integration),
the width of the structure,
and whether the top or bottom strand of the query sequence was being used for the comparison.
When all of the files in the horizontal search set have been examined,
the list of N-best structures is reported,
as shown in the output file above.
The Wisconsin Package must be configured for graphics before you run any program with graphics output! If the % setplot command is available in your installation,
this is the easiest way to establish your graphics configuration,
but you can also use commands like % postscript that correspond to the graphics languages the Wisconsin Package supports.
See Chapter 5,
Using Graphics in the User's Guide
for more information about configuring your process for graphics.
If you need to stop this program,
use
All parameters for this program may be put on the command line.
Use the option -CHEck to see the summary below and to have a chance to add things to the command line before the program executes.
In the summary below,
the capitalized letters in the qualifier names are the letters that you must type in order to use the parameter.
Square brackets ([ and ])
enclose qualifiers or parameter values that are optional.
For more information,
see "Using Program Parameters" in Chapter 3,
Basic Concepts: Using Programs in the GCG User's Guide.
TWordSearch was conceived and written by John Devereux and Paul Haeberli.
TWordSearch uses an algorithm very similar to Wilbur and Lipman (Proc.
Natl.
Acad.
Sci.
(USA)
80;
726-730 (1983)).
We learned about masked word searching in a personal communication from Temple Smith of the Dana Farber Cancer Institute.
The files described below supply auxiliary data to this program.
The program automatically reads them from a public data directory unless you either 1)
have a data file with exactly the same name in your current working directory;
or 2)
name a file on the command line with an expression like -DATa1=myfile.dat.
For more information see Chapter 4,
Using Data Files in the User's Guide.
If you use the command line option -SIMplify,
TWordSearch reads the local data file simplify.txt to find the symbol equivalences you want to use.
You can specify a simplification table with another name on the command line with an expression like -SIMplify=
mysimplify.txt.
There is more on the subject of sequence simplification in the documentation for the Simplify
program.
The simplify.txt file in the public data directory is only appropriate for simplifying peptide sequences.
You must create your own simplify.txt file to define equivalences for nucleic acid simplifications.
The translation of codons to amino acids,
the identification of potential start codons and stop codons,
and the mappings of one-letter to three-letter amino acid codes are all defined in a translation table in the file translate.txt.
If the standard genetic code does not apply to your sequence,
you can provide a modified version of this file in your working directory or name an alternative file on the command line with an expression like -TRANSlate=
mycode.txt.
Translation tables are discussed in more detail in the Data Files manual.
The parameters and switches listed below can be set from the command line.
For more information,
see "Using Program Parameters" in Chapter 3,
Basic Concepts: Using Programs in the GCG User's Guide.
sets a threshold score,
from 1 to 100,
at or below which a diagonal cannot be considered.
makes a plot showing the distribution (frequency)
of diagonal scores.
The score distribution plot is useful for determining if a score in the output list is significant.
You must have a plotter or graphic screen to use this option.
There is a whole paragraph above about the score distribution plot.
simplifies the sequences before comparison according to a table of equivalences in the local data file called simplify.txt (see the LOCAL DATA FILES
topic above).
Many investigators feel that peptide sequence pattern recognition for word searching is more sensitive if similar amino acids are treated as equivalent.
You can name a file other than simplify.txt with the optional parameter.
limits the search to sequences that have been entered into the database or modified since June 1990.
As this is being written,
only the EMBL,
GenBank,
and SWISS-PROT databases support this feature.
If the query sequence really is a protein,
but TWordSearch guesses wrong,
you see the alphabet parameter set equal to 4 and the word "Nucleotide" next to "WordSearch"
at the top of the output list.
The search will probably not find any word matches at all! The command line option -PROtein tells TWordSearch to make the word search based on the alphabet of characters found in the query sequence.
causes TWordSearch to sort the list of diagonals a second time by sequence name,
so that all of the diagonals from the same sequence appear together in the output list.
Usually,
the diagonal list from TWordSearch is shown with the most significant (highest score)
diagonal first and diagonals with successively lower scores following.
While this is the obvious order,
it slows down the TSegments
display program that has to read each sequence in the list to make the display.
Usually,
translation is based on the translation table in a default or local data file called translate.txt.
This option allows you to use a translation table in a different file.
(See the Data Files manual for information about translation tables.)
This program normally monitors its progress on your screen.
However,
when you use the -Default option to suppress all program interaction,
you also suppress the monitor.
You can turn it back on with this option.
If your program is running in batch,
the monitor will appear in the log file.
If the monitor is slowing the program down,
suppress it with -NOMONitor.
writes a summary of the program's work to the screen when you've used the -Default qualifier to suppress all program interaction.
A summary typically displays at the end of a program run interactively.
You can suppress the summary for a program run interactively with -NOSUMmary.
Use this qualifier also to include a summary of the program's work in the log file for a program run in batch.
submits the program to the batch queue for processing after prompting you for all required user inputs.
Any information that would normally appear on the screen while the program is running is written into a log file.
Whether that log file is deleted,
printed,
or saved to your current directory depends on how your system manager has set up the command that submits this program to the batch queue.
All output files are written to your current directory,
unless you direct the output to another directory when you specify the output file.
suppresses the documentation at the end of each line in the output list.
These options apply to all GCG graphics programs.
These and many others are described in detail in Chapter 5,
Using Graphics of the User's Guide.
writes the plot as a text file of plotting instructions suitable for input to the Figure
program instead of drawing the plot on your plotter.
draws all text characters on the plot using Font 3 (see Appendix I)
.
draws the entire plot with the pen in stall 1.
These options let you expand or reduce the plot (zoom),
move it in either direction (pan),
or rotate it 90 degrees (rotate).
expands the plot by 20 percent by resetting the scaling factor (normally 1.0)
to 1.2 (zoom in).
You can expand the axes independently with -XSCAle and -YSCAle.
Numbers less than 1.0 contract the plot (zoom out).
moves the plot to the right by 30 platen units (pan right).
moves the plot up by 30 platen units (pan up).
rotates the plot 90 degrees.
Usually,
plots are displayed with the horizontal axis longer than the vertical (landscape).
Note that plots are reduced or enlarged,
depending on the platen size,
to fill the page.
Wilbur,
W.J.
and Lipman,
D.J.
(1983).
Rapid Similarity Searches of Nucleic Acid and Protein Data Banks.
Proceedings of the National Academy of Sciences USA 80,
726-730.
Printed: April 22,
1996 15:56 (1162)
LIST REFINEMENT
Adding Lists Together
Suppressing Items
OUTPUT FILE FORMAT
The Heading
The List of Best Diagonals
ALGORITHM
Score
Alphabet
The Histogram is Integrated
GRAPHICS
CTRL-C
COMMAND-LINE SUMMARY
Minimum syntax: % twordsearch [-INfile1=]GGammaCod.Seq -Default
Prompted parameters:
-BEGin=1 -END=444 range of interest
[-INfile2=]SW:* Search set (all of Swiss-Prot)
-WORdsize=6 or -MASk=++-++-++ word size or mask pattern
-LIStsize=50 size of output list
-INTegrate=3 width of integration window
[-OUTfile=]ggammacod.word output file name
Local Data Files:
[-SIMplify=]simplify.txt optional simplification table
[-TRANSlate=]translate.txt contains the genetic code
Optional Parameters:
-SIMplify[=fname] simplifies sequences [using table in fname]
-SINce=6.90 limits search to sequences dated on or after June 1990
-NOMONitor suppresses the screen trace for each search set
sequence
-NOSUMmary suppresses the summary of the search
-RESORt sorts output list by name instead of score
-PLOt makes a plot of the score distribution
-LOWscore=10 sets minimum score (from 1 to 100) for diagonal to be
listed
-NOSHOwfiles suppresses documentation at the end of each line in the
output
-BATch submits the program to run in the batch queue
All GCG graphics programs accept these and other switches. See the Using
Graphics chapter of the USERS GUIDE for descriptions.
-FIGure[=FileName] stores plot in a file for later input to FIGURE
-FONT=3 draws all text on the plot using font 3
-COLor=1 draws entire plot with pen in stall 1
-SCAle=1.2 enlarges the plot by 20 percent (zoom in)
-XPAN=10.0 moves plot to the right 10 platen units (pan right)
-YPAN=10.0 moves plot up 10 platen units (pan up)
-PORtrait rotates plot 90 degrees
ACKNOWLEDGEMENT
LOCAL DATA FILES
OPTIONAL PARAMETERS
-LOWscore=10
-PLOt
-SIMplify=fname
-SINce=6.90
-RESORt
-TRANSlate=filename.txt
-MONitor
-SUMmary
-BATch
-NOSHOwfiles
-FIGure=programname.figure
-FONT=3
-COLor=1
-SCAle=1.2
-XPAN=30.0
-YPAN=30.0
-PORtrait
REFERENCES