



/featured3/2015RNA-PuzzlesRoundII_ftr_pic.png) |
RNA-Puzzles Round II: assessment of RNA structure
prediction programs applied to three large RNA structures This paper is a report of a second round of RNA-Puzzles, a collective and blind experiment in three-dimensional (3D) RNA structure prediction. Three puzzles, Puzzles 5, 6, and 10, represented sequences of three large RNA structures with limited or no homology with previously solved RNA molecules. A lariat-capping ribozyme, as well as riboswitches complexed to adenosylcobalamin and tRNA, were predicted by seven groups using RNAComposer, ModeRNA/SimRNA, Vfold, Rosetta, DMD, MC-Fold, 3dRNA, and AMBER refinement. Some groups derived models using data from state-of-the-art chemical-mapping methods (SHAPE, DMS, CMCT, and mutate-and-map). The comparisons between the predictions and the three subsequently released crystallographic structures, solved at diffraction resolutions of 2.5–3.2 Å, were carried out automatically using various sets of quality indicators. The comparisons clearly demonstrate the state of present-day de novo prediction abilities as well as the limitations of these state-of-the-art methods. All of the best prediction models have similar topologies to the native structures, which suggests that computational methods for RNA structure prediction can already provide useful structural information for biological problems. However, the prediction accuracy for non-Watson–Crick interactions, key to proper folding of RNAs, is low and some predicted models had high Clash Scores. These two difficulties point to some of the continuing bottlenecks in RNA structure prediction. All submitted models are available for download at http://ahsoka.u-strasbg.fr/rnapuzzles/.
|
/featured3/Science-2015-Shen-75-8Pic.png) |
Protein synthesis. Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains Abstract: In Eukarya, stalled translation induces 40S dissociation and recruitment of the ribosome quality control complex (RQC) to the 60S subunit, which mediates nascent chain degradation. Here we report cryo-electron microscopy structures revealing that the RQC components Rqc2p (YPL009C/Tae2) and Ltn1p (YMR247C/Rkr1) bind to the 60S subunit at sites exposed after 40S dissociation, placing the Ltn1p RING (Really Interesting New Gene) domain near the exit channel and Rqc2p over the P-site transfer RNA (tRNA). We further demonstrate that Rqc2p recruits alanine- and threonine-charged tRNA to the A site and directs the elongation of nascent chains independently of mRNA or 40S subunits. Our work uncovers an unexpected mechanism of protein synthesis, in which a protein--not an mRNA--determines tRNA recruitment and the tagging of nascent chains with carboxy-terminal Ala and Thr extensions ("CAT tails").
|
/featured3/PfefferGolgiAssociatedWSupp.png) |
Golgi-associated RhoBTB3 targets Cyclin E for ubiquitylation and promotes cell cycle progression Abstract: Cyclin E regulates the cell cycle transition from G1 to S phase and is degraded before entry into G2 phase. Here we show that RhoBTB3, a Golgiassociated,
Rho-related ATPase, regulates the S/G2 transition of the cell cycle by targeting Cyclin E for ubiquitylation. Depletion of RhoBTB3 arrested cells in S phase, triggered Golgi fragmentation, and elevated Cyclin E
levels. On the Golgi, RhoBTB3 bound Cyclin E as part of a Cullin3 (CUL3)-dependent RING–E3 ubiquitin ligase complex comprised of RhoBTB3, CUL3, and RBX1. Golgi association of this complex was required for its ability to
catalyze Cyclin E ubiquitylation and allow normal cell cycle progression. These experiments reveal a novel role for a Ras superfamily member in catalyzing Cyclin E turnover during S phase, as well as an unexpected, essential
role for the Golgi as a ubiquitylation platform for cell cycle control.
|
/featured3/SalzmanFeaturedlabpic.png) |
Cell-Type Specific Features of Circular RNA Expression Abstract: Thousands of loci in the human and mouse genomes give rise to circular RNA transcripts; at many of these loci, the predominant RNA isoform is a circle. Using an improved computational approach for circular RNA identification, we found widespread circular RNA expression in Drosophila melanogaster and estimate that in humans, circular RNA may account for 1% as many molecules as poly(A) RNA. Analysis of data from the ENCODE consortium revealed that the repertoire of genes expressing circular RNA, the ratio of circular to linear transcripts for each gene, and even the pattern of splice isoforms of circular RNAs from each gene were cell-type specific. These results suggest that biogenesis of circular RNA is an integral, conserved, and regulated feature of the gene expression program.
|
/featured3/yehchemicalrescuepic2b.png) |
Chemical Rescue of Malaria Parasites Lacking an Apicoplast Defines Organelle Function in Blood-Stage Plasmodium falciparum
Abstract: Plasmodium spp parasites harbor an unusual plastid organelle called the apicoplast. Due to its prokaryotic origin and essential function, the apicoplast is a key target for development of new anti-malarials. Over 500 proteins are predicted to localize to this organelle and several prokaryotic biochemical pathways have been annotated, yet the essential role of the apicoplast during human infection remains a mystery. Previous work showed that treatment with fosmidomycin, an inhibitor of non-mevalonate isoprenoid precursor biosynthesis in the apicoplast, inhibits the growth of blood-stage P. falciparum. Herein, we demonstrate that fosmidomycin inhibition can be chemically rescued by supplementation with isopentenyl pyrophosphate (IPP), the pathway product. Surprisingly, IPP supplementation also completely reverses death following treatment with antibiotics that cause loss of the apicoplast. We show that antibiotic-treated parasites rescued with IPP over multiple cycles specifically lose their apicoplast genome and fail to process or localize organelle proteins, rendering them functionally apicoplast-minus. Despite the loss of this essential organelle, these apicoplast-minus auxotrophs can be grown indefinitely in asexual blood stage culture but are entirely dependent on exogenous IPP for survival. These findings indicate that isoprenoid precursor biosynthesis is the only essential function of the apicoplast during blood-stage growth. Moreover, apicoplast-minus P. falciparum strains will be a powerful tool for further investigation of apicoplast biology as well as drug and vaccine development.
|
/featured3/herschlag.jpg) |
Using Unnatural Amino Acids to Probe the Energetics of Oxyanion Hole Hydrogen Bonds in the Ketosteroid Isomerase Active Site
Abstract: Hydrogen bonds are ubiquitous in enzyme active sites, providing binding interactions and stabilizing charge rearrangements on substrate groups over the course of a reaction. But understanding the origin and magnitude of their catalytic contributions relative to hydrogen bonds made in aqueous solution remains difficult, in part because of complexities encountered in energetic interpretation of traditional site-directed mutagenesis experiments. It has been proposed for ketosteroid isomerase and other enzymes that active site hydrogen bonding groups provide energetic stabilization via “short, strong” or “low-barrier” hydrogen bonds that are formed due to matching of their pKa or proton affinity to that of the transition state. It has also been proposed that the ketosteroid isomerase and other enzyme active sites provide electrostatic environments that result in larger energetic responses (i.e., greater “sensitivity”) to ground-state to transition-state charge rearrangement, relative to aqueous solution, thereby providing catalysis relative to the corresponding reaction in water. To test these models, we substituted tyrosine with fluorotyrosines (F-Tyr’s) in the ketosteroid isomerase (KSI) oxyanion hole to systematically vary the proton affinity of an active site hydrogen bond donor while minimizing steric or structural effects. We found that a 40-fold increase in intrinsic F-Tyr acidity caused no significant change in activity for reactions with three different substrates. F-Tyr substitution did not change the solvent or primary kinetic isotope effect for proton abstraction, consistent with no change in mechanism arising from these substitutions. The observed shallow dependence of activity on the pKa of the substituted Tyr residues suggests that the KSI oxyanion hole does not provide catalysis by forming an energetically exceptional pKa-matched hydrogen bond. In addition, the shallow dependence provides no indication of an active site electrostatic environment that greatly enhances the energetic response to charge accumulation, consistent with prior experimental results.
|
 |
TPP1 OB-Fold Domain Controls Telomere Maintenance by Recruiting Telomerase to Chromosome Ends
Abstract: Telomere synthesis in cancer cells and stem cells involves trafficking of telomerase to Cajal bodies, and telomerase is thought to be recruited to telomeres through interactions with telomere-binding proteins. Here, we show that the OB-fold domain of the telomere-binding protein TPP1 recruits telomerase to telomeres through an association with the telomerase reverse transcriptase TERT. When tethered away from telomeres and other telomerebinding proteins, the TPP1 OB-fold domain is sufficient to recruit telomerase to a heterologous chromatin locus. Expression of a minimal TPP1 OBfold inhibits telomere maintenance by blocking access of telomerase to its cognate binding site at telomeres. We identify amino acids required for the TPP1-telomerase interaction, including specific loop residues within the TPP1 OB-fold domain and individual residues within TERT, some of which are mutated in a subset of pulmonary fibrosis patients. These data define a potential interface for telomerase-TPP1 interaction required for telomere maintenance and implicate defective telomerase recruitment in telomerase-related disease.
|
 |
Structural ensemble and microscopic elasticity of freely diffusing DNA by direct measurement of fluctuations
Abstract: Precisely measuring the ensemble of conformers that a macromolecule populates in solution is highly challenging. Thus, it has been difficult to confirm or falsify the predictions of nanometer-scale dynamical modeling. Here, we apply an X-ray interferometry technique to probe the solution structure and fluctuations of B-form DNA on a length scale comparable to a protein-binding site. We determine an extensive set of intrahelix distance distributions between pairs of probes placed at distinct points on the surface of the DNA duplex. The distributions of measured distances reveal the nature and extent of the thermally driven mechanical deformations of the helix. We describe these deformations in terms of elastic constants, as is common for DNA and other polymers. The average solution structure and microscopic elasticity measured by X-ray interferometry are in striking agreement with values derived from DNA–protein crystal structures and measured by force spectroscopy, with one exception. The observed microscopic torsional rigidity of DNA is much lower than is measured by single-molecule twisting experiments, suggesting that torsional rigidity increases when DNA is stretched. Looking forward, molecular-level interferometry can provide a general tool for characterizing solution-phase structural ensembles.
|
|
Genomic responses in mouse models poorly mimic human inflammatory diseases.
Abstract: A cornerstone of modern biomedical research is the use of mouse models to explore basic pathophysiological mechanisms, evaluate new therapeutic approaches, and make go or no-go decisions to carry new drug candidates forward into clinical trials. Systematic studies evaluating how well murine models mimic human inflammatory diseases are nonexistent. Here, we show that, although acute inflammatory stresses from different etiologies result in highly similar genomic responses in humans, the responses in corresponding mouse models correlate poorly with the human conditions and also, one another. Among genes changed significantly in humans, the murine orthologs are close to random in matching their human counterparts (e.g., R2 between 0.0 and 0.1). In addition to improvements in the current animal model systems, our study supports higher priority for translational medical research to focus on the more complex human conditions rather than relying on mouse models to study human inflammatory diseases. PRESS COVERAGE
|
|
Radial Construction of an Arterial Wall Abstract: Some of the most serious diseases involve altered size and structure of the arterial wall. Elucidating how arterial walls are built could aid understanding of these diseases, but little is known about how concentric layers of muscle cells and the outer adventitial layer are assembled and patterned around endothelial tubes. Using histochemical, clonal, and genetic analysis in mice, here we show that the pulmonary artery wall is constructed radially, from the inside out, by two separate but coordinated processes. One is sequential induction of successive cell layers from surrounding mesenchyme. The other is controlled invasion of outer layers by inner layer cells through developmentally regulated cell reorientation and radial migration. We propose that a radial signal gradient controls these processes and provide evidence that PDGF-B and at least one other signal contribute. Modulation of such radial signaling pathways may underlie vessel-specific differences and pathological changes in arterial wall size and structure.
|
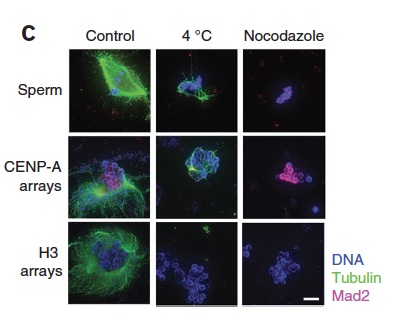 |
A cell-free system for functional centromere and kinetochore assembly Abstract: This protocol describes a cell-free system for studying vertebrate centromere and kinetochore formation. We reconstitute tandem arrays of centromere protein A (CENP-A) nucleosomes as a substrate for centromere and kinetochore assembly. These chromatin substrates are immobilized on magnetic beads and then incubated in Xenopus egg extracts that provide a source for centromere and kinetochore proteins and that can be cycled between mitotic and interphase cell cycle states. This cell-free system lends itself to use in protein immunodepletion, complementation and drug inhibition as a tool to perturb centromere and kinetochore assembly, cytoskeletal dynamics, DNA modification and protein post-translational modification. This system provides a distinct advantage over cell-based investigations in which perturbing centromere and kinetochore function often results in lethality. After incubation in egg extract, reconstituted CENP-A chromatin specifically assembles centromere and kinetochore proteins, which locally stabilize microtubules and, on microtubule depolymerization with nocodazole, activate the mitotic checkpoint.
|
|
Imaging nanometre-scale structure in cells using in situ aberration correction
Abstract: Accurate distance measurements of cellular structures on a length scale relevant to single macromolecules or macromolecular complexes present a major challenge for biological microscopy. In addition to the inherent challenges of overcoming the limits imposed by the diffraction of light, cells themselves are a complex and poorly understood optical environment. We present an extension of the high-resolution colocalization method to measure three dimensional distances between diffraction-limited objects using standard widefield fluorescence microscopy. We use this method to demonstrate that in three dimensions, cells intrinsically introduce a large and variable amount of chromatic aberration into optical measurements. We present a means of correcting this aberration in situ [termed ‘Colocalization and In-situ Correction of Aberration for Distance Analysis’ (CICADA)] by exploiting the fact that there is a linear relationship between the degree of aberration between different wavelengths. By labelling a cellular structure with redundantly multi-colour labelled antibodies, we can create an intracellular fiducial marker for correcting the individual aberrations between two different wavelengths in the same cells. Our observations demonstrate that with suitable corrections, nanometre scale three-dimensional distance measurements can be used to probe the substructure of macromolecular complexes within cells. |
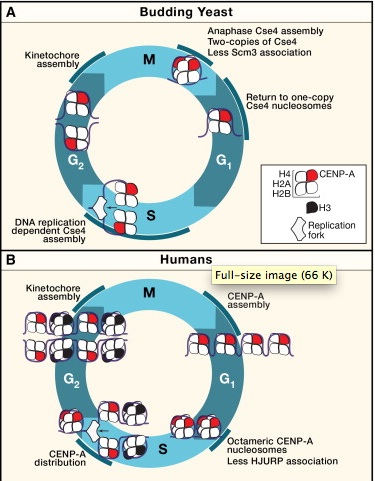 |
The split personality of CENP-A nucleosomes
A specialized chromatin domain, called the centromere, ensures accurate chromosome segregation during mitosis. Centromeres are the foundation for the assembly of the kinetochore, the site on each chromosome that acts as the primary interface between the chromosomes and the microtubules of the mitotic spindle. Maintaining the centromere is therefore essential for chromosome stability. The chromatin mark that determines centromere identity is a specialized histone H3 variant called CENP-A (called Cse4 in the budding yeast Saccharomyces cerevisiae). Budding yeast assemble Cse4 into chromatin during each round of DNA replication, whereas vertebrate CENP-A nucleosome assembly is replication independent, occurring during telophase and G1. |


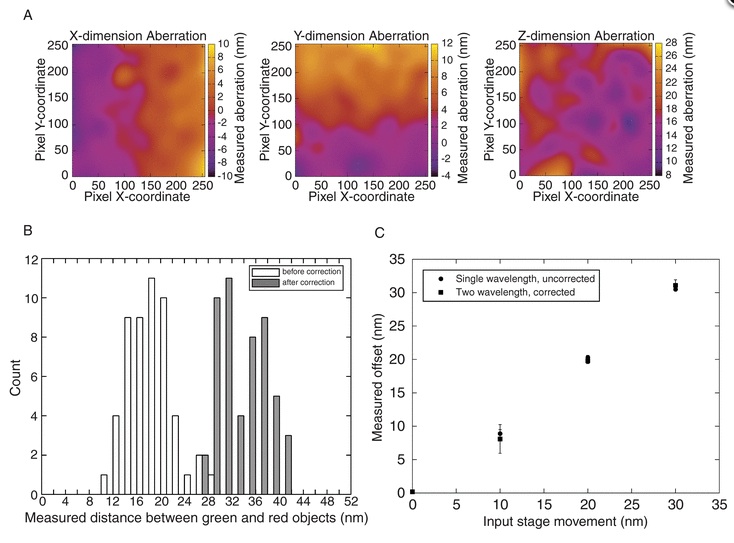

| |